晶型药物实验室已完成或在研部分课题
晶型药物实验室已完成或在研部分课题
山东省晶型药物工程技术研究中心对国内临床用药需求急迫、社会需求量大的品牌药物进行创新性研究,开发高水平的仿制药、有特色的创新药物是本课题的重要工作。从晶型药物研究出发,针对市场需求量较大的药物种类寻找其优势晶型,提高原料药质量控制,增强制剂稳定性和生物利用度,提高临床用药质量和疗效水平。本中心目前正在与中国医学科学院药物研究所、山东大学合作成功开发了国家一类新药新尼群地平、硫酸氢氯吡格雷优势晶型、新葛根素优势晶型、罗红霉素优势晶型药物、泮托拉唑优势晶型药物、左氧氟沙星优势晶型等10余项创新晶型药物,已完成临床前研究或获得新药临床批件,掌握了晶型药物的关键技术和核心内容,已申请国家发明专利7项,现将研究的主要内容介绍如下:
一、相关研究课题
(1) 葛根素优势晶型药物
研究背景
本研究基于已获得的国家发明专利“葛根素一种新的优势药用晶型固体物质及制备方法与药物组合物在临床中的应用”。
葛根素分子结构如图所示:
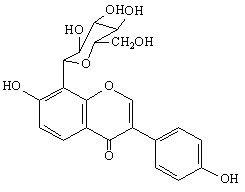
本发明目的是从葛根素化合物的固体存在状态研究入手,通过晶型筛选技术,在药物有效成分的原料层面上寻找、发现葛根素晶型固体物质存在种类与状态特征,将晶型研究与药效学研究相结合,寻找、发现、开发适合固体给药途径的具有最佳临床疗效的葛根素晶型固体物质状态,为晶型固体药物开发提供基础的科学研究数据;同时,也为从葛根素固体药物原料物质基础上申请国家或国际的知识产权发明专利保护提供科学依据。
前期研究基础
本发明发现了葛根素一种优势新晶V型固体物质存在形式;发明了生产葛根素晶V型固体物质的制备方法;涉及发现晶V型物质在生物体内具有吸收优势和最大血药浓度的长久持续平台期特性;涉及采用这种新晶型物质作为药物活性成分制备开发出的药物组合物在防治心脑血管疾病,特别对扩张冠状动脉、改善心脑血管微循环等具有明显的优势临床治疗作用。
研究目标
本研究在已获得的国家发明专利基础上,进行葛根素优势晶型固体药物开发。
①进行葛根素优势晶型固体药物原料研究;
②进行葛根素优势晶型固体制剂研究和质控研究;
③完成优势晶型固体药物的临床前和临床研究,提升药物产品质量水平;
④推进产业化进程,完善晶型药物研究相关国家标准。
(2) 罗红霉素优势晶型药物
研究背景
本研究基于已获得的国家发明专利“罗红霉素化合物的三种晶型固体物质与制备方法及其药物组合物在临床中的应用”。
罗红霉素分子结构如图所示:
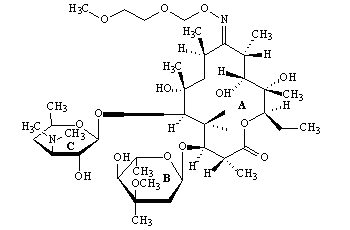
本发明发现了罗红霉素化合物有晶A型、晶B型、晶C型三种固体物质存在形式;发明了生产罗红霉素三种晶型固体样品的制备方法;发现使用罗红霉素不同晶型物质作为活性成分制备开发出的各种药物制剂及药物组合在金黄色葡萄球菌、链球菌、棒状杆菌、李司忒菌、卡他摩拉菌、军团菌等敏感菌所致的呼吸道、泌尿道、皮肤和软组织、五官科感染等疾病中发挥的优势临床治疗作用疗效不同;发现晶型影响罗红霉素固体药物有效成分在生物体内的吸收速度、增强或减少生物体内血药浓度从而影响药物在临床中的疗效作用。
本发明目的之一:是提供不含结晶水或其它结晶溶剂的罗红霉素晶A型、晶B型与晶C型的三种固体物质存在状态和描述方式。
本发明目的之二:是提供罗红霉素晶A型、晶B型、晶C型的三种固体物质样品的制备方法。
本发明目的之三:是提供含有使用罗红霉素晶型物质(含晶A型、或晶B型、或晶C型、或由两种及三种晶型成分组成的混合晶型物质)作为药物活性成分原料制备开发出的药物组合物在防治各类感染类疾病中发挥的优势临床治疗作用差异。
本发明目的之四:是提供使用含有罗红霉素晶型物质(含晶A型、或晶B型、或晶C型、或由两种及三种晶型成分组成的混合晶型物质)作为药物活性成分的人体每日给药剂量范围。
本发明目的之五:是提供使用含有罗红霉素晶型物质(含晶A型、或晶B型、或晶C型、或由两种及三种晶型成分组成的混合晶型物质)作为药物活性成分原料而制备开发出供临床使用的各种片剂、胶囊、丸剂、针剂、缓释或控释各种药物制剂及药物组合物。
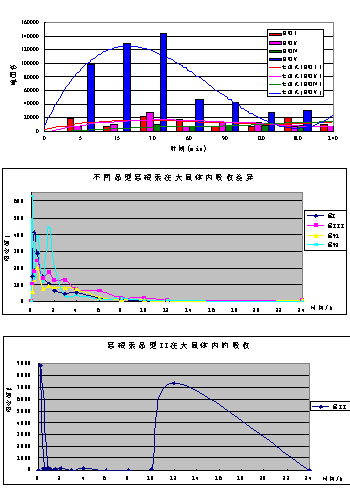
本发明目的之六:是提供罗红霉素三种晶型样品在生物体内存在的吸收和血药浓度差异数据,晶A型较晶B型、晶C型吸收好,在1小时可达最大浓度,表明罗红霉素的晶A型更易于通过胃肠道吸收而发挥较好的临床疗效作用。
前期研究
我们的研究显示罗红霉素化合物有晶A型、晶B型、晶C型三种固体物质存在形式(见下图)。
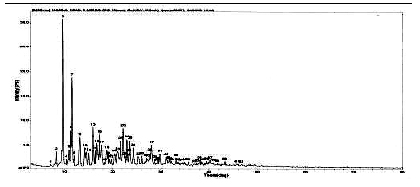
图 罗红霉素晶A型的X衍射分析图谱
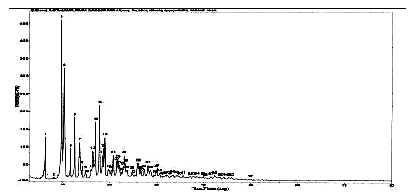
图 罗红霉素晶B型的X衍射分析图谱
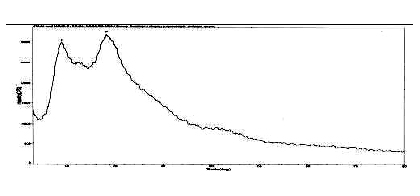
图 罗红霉素晶C型的X衍射分析图谱
大鼠体内药代动力学研究结果发现晶型影响罗红霉素固体药物有效成分在生物体内的吸收速度、增强或减少生物体内血药浓度从而影响药物在临床中的疗效作用。
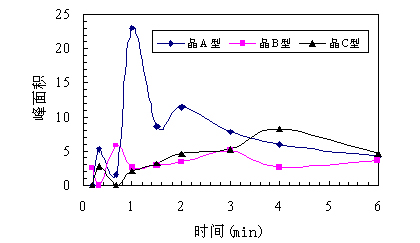
图 罗红霉素三种晶型样品在大鼠体内存在的吸收和血药浓度
晶A型较晶B型、晶C型吸收好,在1小时可达最大浓度,表明罗红霉素的晶A型更易于通过胃肠道吸收而发挥较好的临床疗效作用
研究目标
本研究在已获得的国家发明专利基础上,进行罗红霉素优势晶型固体药物原料药和制剂开发。
①进行罗红霉素优势晶型固体药物原料研究;
②进行罗红霉素优势晶型固体制剂研究和质控研究;
③完成罗红霉素优势晶型固体药物的临床前和临床研究,提升药物质量水平;
④推进产业化进程,完善晶型药物研究相关国家标准。
(3) 阿托伐他汀
研究背景
阿托伐他汀为HMG-CoA还原酶选择性抑制剂,通过抑制HMG-CoA还原酶和胆固醇在肝脏的生物合成而降低血浆胆固醇和脂蛋白水平,并能通过增加肝细胞表面低密度脂蛋白(LDL)受体数目而增加LDL的摄取和分解代谢。本品也能减少LDL的生成和其颗粒数。本品还能降低某些纯合子型家族性高胆固醇血症(FH)患者的低密度脂蛋白胆固醇(LDL-C)水平,而这一类型的人群对其他类型的降脂药物治疗很少有应答。
阿托伐他汀能降低纯合子和杂合子家族性高胆固醇血症、非家族性高胆固醇血症以及混合性脂类代谢障碍患者的血浆总胆固醇(TC)、LDL-C和载脂蛋白B(ApoB),还能降低极低密度脂蛋白胆固醇(VLDL-C)和三酰甘油(TG)的水平,并能不同程度地提高血浆高密度 脂蛋白胆固醇(HDL-C)和载脂蛋白A1(ApoA1)的水平。
阿托伐他汀是全球处方量最多的降胆固醇药物和全球销售额最大的处方药,它可以显著降低低密度脂蛋白胆固醇和甘油三酯水平,并能够显著降低高血压患者、糖尿病患者、冠心病患者心肌梗塞和卒中等主要心脑血管事件发生的风险。自199年在全球上市以来,其卓越的疗效和良好的安全性在400多个临床试验和1.27亿/年病人治疗的临床用药经验中得到验证。在我国1999年即由北京红惠制药强仿上市。2006医药经济报将阿托伐他汀项目评价为“最具开发价值的药品”,2007年行政保护到期后,国内已有多家企业申报。
前期研究
我们的实验室研究发现了阿托伐他汀化合物以α晶型、β晶型两种固体物质存在形式,发现晶型状态的不同影响阿托伐他汀固体药物有效成分在生物体内的吸收速度、增强或减少生物体内血药浓度从而影响药物在临床中的疗效作用。
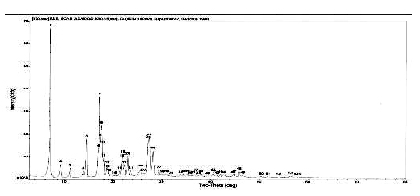
图 阿托伐他汀α晶型的X衍射分析图谱
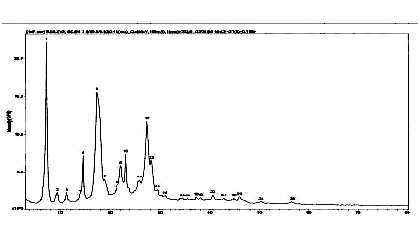
图 阿托伐他汀β晶型的X衍射分析图谱
表 阿托伐他汀不同晶型状态对其体内吸收的影响
|
时间 晶型
|
10 min
|
20 min
|
30 min
|
40 min
|
|
β晶型
|
13.2
|
48.6
|
14.6
|
14.2
|
|
(α+β) 晶型
|
8.5
|
15.6
|
7.9
|
7.1
|
|
α晶型
|
3.8
|
9.7
|
4.4
|
3.5
|
|
α晶型
|
3.2
|
8.0
|
2.2
|
2.4
|
研究目标
通过对阿托伐他汀固体晶型化学物质与其临床疗效的关系,阐明化学药物晶型对生物活性的重要影响作用,并选取优势药用晶型进行开发。
① 寻找、发现、确定阿托伐他汀优势药用晶型;
② 建立对阿托伐他汀固体晶型化学物质的有效鉴别与质量控制分析方法;
③ 按照SFDA新药申报要求,完成阿托伐他汀新优势晶型药物的全部临床前研究、临床研究;
④ 为我国非专利的晶型药物的质量控制提供引导研究的示范作用。
(4) 左氧氟沙星
研究背景
世界抗感染药物市场重心正由头孢菌素类药物向氟喹诺酮类药物转移,氟喹诺酮类药物成为抗生素药物中发展最快的领域。相对于头孢类等抗感染药物,氟喹诺酮(fluoroquinolone)类药物获得了价格低、药效高等比较优势;因此,为了追求更好的疗效和口服生物利用度,医疗人员越来越倾向于使用氟喹诺酮类和其他类别的抗菌素。左氧氟沙星是目前抗菌药市场上的明星品种,左氧氟沙星于1997年在加拿大上市,是一种氟喹诺酮类广谱抗生素,用于治疗成人呼吸道、皮肤及尿道细菌感染。2003年居国内医院抗菌素用药的榜首,我国目前年产左氧氟沙星40余吨。
氟喹诺酮类抗菌剂在临床上对于泌尿生殖系统感染、呼吸系统感染、消化系统炎症、妇科炎症、性病以及皮肤和软组织的感染均有比较好的效果,近年来发展快速。据预测,未来5年内,世界氟喹诺酮药物市场将迎来最快的增长阶段,对氟喹诺酮原料药的需求也将呈现高速增长。在氟喹诺酮领域,我国的优势企业已经在相应领域建立了自己的产品优势。他们各自拥有自己的产品优势,在原料药的基础上,延伸制剂产品链,从而获得最大竞争优势。
前期研究
我们的实验室研究发现了左氧氟沙星化合物有晶A型、晶B型、晶C型三种固体物质存在形式,发现晶型状态的不同影响左氧氟沙星固体药物有效成分在生物体内的吸收速度、增强或减少生物体内血药浓度从而影响药物在临床中的疗效作用。
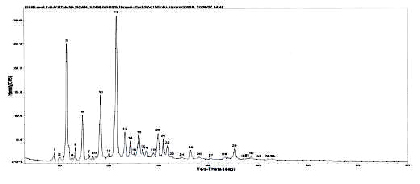
图 左氧氟沙星晶A型的X衍射分析图谱
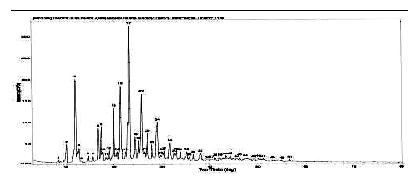
图 左氧氟沙星晶B型的X衍射分析图谱
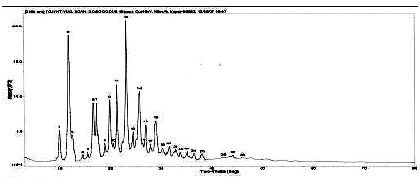
图 左氧氟沙星晶C型的X衍射分析图谱
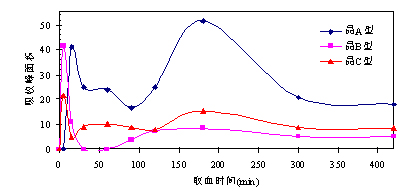
图 左氧氟沙星三种晶型样品在大鼠体内存在的吸收和血药浓度
研究目标
通过对左氧氟沙星固体晶型化学物质与其临床疗效的关系,阐明化学药物晶型对生物活性的重要影响作用,并选取优势药用晶型进行开发。
①寻找、发现、确定左氧氟沙星优势药用晶型;
②建立对左氧氟沙星固体晶型化学物质的有效鉴别与质量控制分析方法;
③按照SFDA新药申报要求,完成左氧氟沙星新优势晶型药物的全部临床前研究、临床研究;
④为我国非专利的晶型药物的质量控制提供引导研究的示范作用。
(5) 泮托拉唑
研究背景
浸蚀性食道炎是GORD(胃-食道返流性疾病)中的一种严重性慢性病,如果不加治疗会并发更严重症状如胃溃疡、食道狭窄、出血、食道癌及Barrett氏食道病。泮托拉唑缓释片剂剂型属于质子泵抑制剂,用于治疗侵蚀性食道炎,疗程16周。
泮托拉唑是苯并咪唑衍生物,通过与胃壁纤维的质子泵的特异性综合、抑制胃酸分泌。泮托拉唑是壁细胞酸性环境下被激活成活性形式,抑制H+、K+-ATP酶,即胃酸分泌的最终环节。抑制呈剂量依赖关系,并且影响胃酸的基础酸分泌和最大酸分泌。与其他质子泵抑制齐和H2受体拮抗剂一样,使用泮托拉唑治疗后,胃酸分泌减少而胃泌素水平随胃酸减少相应升高。这种胃泌素水平升高是可逆的。因为泮托拉唑在细胞受体水平上与酶的远端结合,因此泮托拉唑可独立或在其他物质(乙酰胆碱、组织胺、胃泌素)的刺激下影响胃酸分泌。口服和静脉使用泮托拉唑,可达到相同的效果。药理作用:毒理作用 急性毒理学研究表明,人鼠静脉应用本品后半数致死量(LD50)为390mg/kg,小鼠为250mg/kg。慢性毒理学研究显示,泮托拉唑可引起动物(大鼠、小鼠)血液中胃泌毒水平上升,并导致胃粘膜形态改变和胃重量增加,这种效应具有可逆性,随用药终止可自然消失。本品不影响生育,亦无致畸的证据。
前期研究
我们的实验室研究发现了泮托拉唑化合物有晶态、非晶态两种固体物质存在形式,发现晶型状态的不同影响泮托拉唑固体药物有效成分在生物体内的吸收速度、增强或减少生物体内血药浓度从而影响药物在临床中的疗效作用。
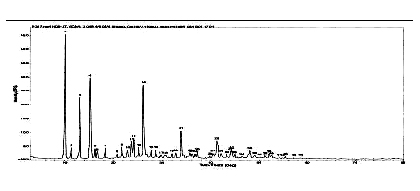
图 晶态泮托拉唑的X衍射分析图 谱
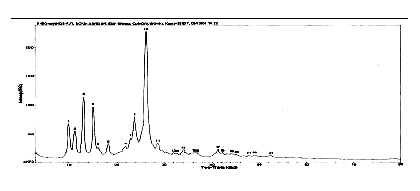
图 非晶态泮托拉唑的X衍射分析图谱
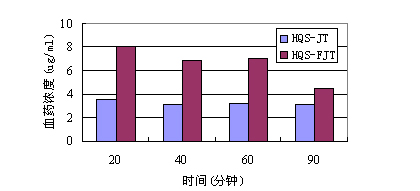
图 泮托拉唑不同晶态形式样品在大鼠体内存在的吸收和血药浓度
研究目标
通过对泮托拉唑固体晶型化学物质与其临床疗效的关系,阐明化学药物晶型对生物活性的重要影响作用,并选取优势药用晶型进行开发。
①寻找、发现、确定泮托拉唑优势药用晶型;
②建立对泮托拉唑固体晶型化学物质的有效鉴别与质量控制分析方法;
③按照SFDA新药申报要求,完成泮托拉唑新优势晶型药物的全部临床前研究、临床研究;
④为我国非专利的晶型药物的质量控制提供引导研究的示范作用。
(6) 阿那曲唑
研究背景
阿那曲唑(anastrozole)是第一种既可用于基础辅助治疗-----直接用于乳癌术后---也可作为Tamoxifen跟进疗法的芳香酶抑制剂。一项全球规模最大、随访时间最长的芳香化酶抑制剂乳腺癌辅助治疗研究临床试验得出最新结果:与传统药物三苯氧胺相比,阿那曲唑可以全面有效降低乳腺癌复发和转移风险,延长患者无病生存期;即使在完成规范疗程4年后,阿那曲唑的保护效果仍可持续。这标志着阿那曲唑将成为乳腺癌内分泌辅助治疗的标准方案。
乳腺癌手术后常用的传统内分泌药物是三苯氧胺。三苯氧胺有不少副作用,如可引起子宫内膜癌、血栓栓塞等。该项临床试验对比了第三代芳香化酶抑制剂阿那曲唑与三苯氧胺的效果。试验共有21个国家381个中心参加,是全球多中心临床试验,从1996年起一共有9366位病人参加了试验,随访时间是100个月。阿那曲唑组有25.8%的病人复发,三苯氧胺组病人的复发率是29.9%,阿那曲唑比三苯氧胺降低了4.1%的复发率,同时还降低了远处转移的风险。在安全性上阿那曲唑也优于三苯氧胺。
阿那曲唑是整整五年治疗期中唯一具有成熟安全与耐药性数据的芳香化酶抑制剂,与他莫昔芬相比具有正向的风险效益比。试验结果进一步支持了该专家小组的发现,为芳香化酶抑制剂作为绝经后早期激素敏感性乳腺癌患者的首选治疗奠定了证据基础。
前期研究
我们的实验室研究发现了阿那曲唑化合物有晶A型、晶B型、晶C型三种固体物质存在形式,发现晶型状态的不同影响阿那曲唑固体药物有效成分在生物体内的吸收速度、增强或减少生物体内血药浓度从而影响药物在临床中的疗效作用。
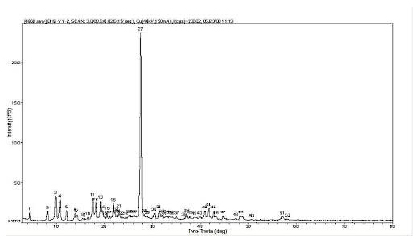
图 阿那曲唑晶A型的X衍射分析图谱
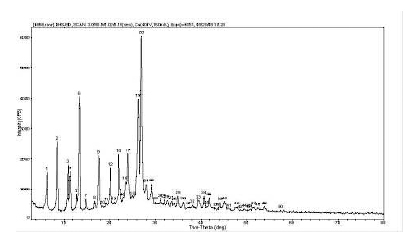
图 阿那曲唑晶B型的X衍射分析图谱
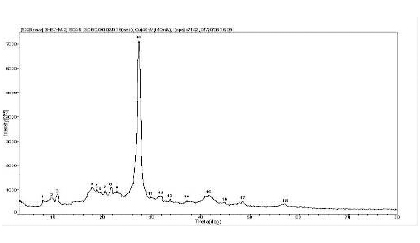
图 阿那曲唑晶C型的X衍射分析图谱
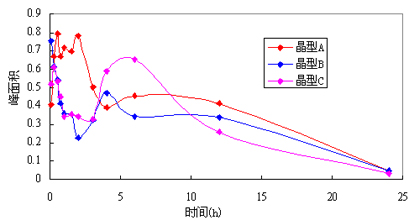
图 阿那曲唑三种晶型样品在大鼠体内存在的吸收
(7)新尼群地平及其制剂
研究背景
尼群地平片2009年列入国家基本药物目录,但国产尼群地平疗效不稳定,不仅不同厂家之间的产品疗效相差甚远,而且同一厂家不同批号产品的疗效和不良反应也存在显著的差异。尼群地平的临床表现直接影响了医生和病人的信心,成为典型的质量不可控制的国产药物。近年来,尽管尼群地平市场价格低廉,除了比较贫困的病人外,该药的应用在逐渐减少,不仅增加了病人和社会的经济负担,而且对病人也形成了直接危害。因此,解决国产尼群地平的质量问题,实现保证药物疗效的质量控制之目的,是急需解决的具有重要社会效益和经济效益的科学问题。
本课题经过长期探索和研究,证明国产尼群地平疗效不稳定的主要原因是固体制剂的尼群地平存在的晶型不同,而在我国尼群地平的质量标准中,目前还没有对尼群地平晶型的质量控制要求,药物生产厂家也没有合理生产工艺保证尼群地平晶型的稳定性。
技术方案
项目拟通过现代药物化学、生物学、药物分析学等交叉学科的技术研究,实现新晶型尼群地平药物的研制与开发最终研究目标。
关键技术
采用物理力学晶格破坏和分子重排专晶方法或通过改变物理的压力条件、温度条件制备获得尼群地平新晶型(IV)固体物质即新尼群地平。
创新之处:1、新的药物晶型,填补国内空白。
2、尼群地平的化合物新晶型-新晶(IV)型固体物质的制备方法填补国内空白。
3、该项目的临床与产业化研究,在国内处于领先水平
阶段性成果
项目已完成所有临床前研究工作,建立成熟的尼群地平优势晶型药物生产工艺,目前已申请生产批件,并进入国家食品药品监督管理局新药审评绿色通道。尼群地平的一种优势晶型、其制法和其药物组合物与用途已申请国家发明专利,申请号:200810102728.X,目前已进入公示期。项目的研制成功将成为我国第一个通过晶型研究获得的国家一类新药。
二、部分课题专利申请书
一种晶I型氯吡格雷硫酸盐的制备方法
1、一种晶I型氯吡格雷硫酸盐的制备方法,其特征在于,包括以下步骤:
(1)在冰水冷却条件或室温条件下,将氯吡格雷硫酸氢盐溶解于正丙醇中,加热溶解并过滤得到澄清溶液;
(2)往步骤(1)得到的澄清溶液中加入醚类沉淀溶剂,于20~37℃条件下搅拌6~9小时,得混悬溶液;
(3)将步骤(2)得到的混悬溶液过滤,得到固体样品,经干燥,得到晶I型氯吡格雷硫酸氢盐。
2、根据权利要求1所述的晶I型氯吡格雷硫酸盐的制备方法,其特征在于,所述步骤(1)中氯吡格雷硫酸氢盐的质量与正丙醇的体积比为1:2~1:20。
3、根据权利要求1所述的晶I型氯吡格雷硫酸盐的制备方法,其特征在于,所述步骤(2)中的醚类沉淀溶剂选自乙醚、叔丁基甲基醚或异丙醚。
4、根据权利要求3所述的晶I型氯吡格雷硫酸盐的制备方法,其特征在于,所述步骤(2)中的醚类沉淀溶剂为乙醚。
5、根据权利要求1所述的晶I型氯吡格雷硫酸盐的制备方法,其特征在于,所述步骤(3)中的干燥温度为40~60℃,干燥时间为10~12小时。
6、一种晶I型氯吡格雷硫酸盐的制备方法,其特征在于,包括以下步骤:
(1)在室温条件下,将氯吡格雷硫酸氢盐溶解于醇类有机溶剂中,加热溶解并过滤得到饱和澄清溶液;
(2)往步骤(1)得到的饱和澄清溶液中加入晶I型氯吡格雷硫酸氢盐晶种,通过搅拌或超声破碎晶种得到混悬溶液;
(3)往步骤(2)得到的混悬溶液中加入沉淀溶剂,经静置,过滤得到固体样品,在40~60℃干燥,得到晶I型氯吡格雷硫酸氢盐。
7、根据权利要求6所述的晶I型氯吡格雷硫酸盐的制备方法,其特征在于,所述步骤(1)中的醇类有机溶剂选自乙醇、正丙醇、乙腈、正丁醇或异丙醇。
8、根据权利要求6所述的晶I型氯吡格雷硫酸盐的制备方法,其特征在于,所述步骤(1)中氯吡格雷硫酸氢盐的质量与醇类有机溶剂的体积比为1:2~1:20。
9、根据权利要求6所述的晶I型氯吡格雷硫酸盐的制备方法,其特征在于,所述步骤(3)中使用的沉淀溶剂选自乙醚、石油醚、正己烷或环己烷。
晶I型氯吡格雷硫酸氢盐的制备方法
技术领域
本发明涉及两种晶I型氯吡格雷硫酸氢盐的制备方法。
背景技术
氯吡格雷硫酸氢盐是一种抗血栓形成药,临床上用于预防和治疗因血小板高聚集状态引起的心、脑及其它动脉循环障碍疾药。
氯吡格雷的化学命名为(+)-(S)-α-(邻氯苯基)-6,7-二氢噻吩并[3,2-c]吡啶-5(4H)-乙酸甲酯,比旋度约为+56°。硫酸氢氯吡格雷的结构式如下图所示:
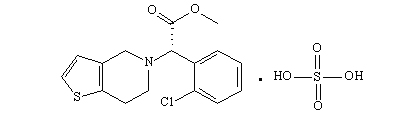
氯吡格雷硫酸氢盐为氯吡格雷的硫酸氢盐,最早由欧洲专利EP 281459推出,是血小板聚集抑制剂,能选择性地抑制ADP与血小板受体的结合,随后抑制激活ADP与糖蛋白GPIIb/IIIa复合物,从而抑制血小板的聚集。可用于防治心肌梗死,缺血性脑血栓,闭塞性脉管炎和动脉粥样硬化及血栓栓塞引起的并发症。
国际公开号WO 99/65915公开了氯吡格雷硫酸氢盐的两种多晶型物质状态,称为形态I和形态II,以及其制备方法。中国专利授权公告号CN 1128805C名称为“氯吡格雷硫酸氢盐的多晶型物及其制备方法和药物组合物”中,涉及了晶I型、晶II型及其制备方法。在中国专利CN 1690060A(公开号)公开了一种制备I型硫酸氯吡格雷的方法,其先得到游离的氯吡格雷碱,再将得到的氯吡格雷游离碱溶解于醚或酯中冷却至-20~5℃,滴加浓硫酸,滴加完毕在-20~20℃搅拌10小时得到I型硫酸氯吡格雷。
以上专利采用的晶I型制备方法可以总结为:得到氯吡格雷游离碱;向氯吡格雷游离碱中加入醚类或酯类试剂,然后向其中滴加硫酸,滴加完毕静置结晶,过滤出晶体即得到晶I型氯吡格雷硫酸氢盐。该方法制备工艺复杂,需要使用惰性气体与并通过滴定硫酸调节,增加了的晶型制备难度。
在中国专利CN1923835A中记载了“氯吡格雷硫酸氢盐的多晶型”。其中,涉及了通过将硫酸氢氯吡格雷盐溶于甲醇和乙醇的溶液中然后加入反溶剂脂肪醚类溶剂混悬制得晶I型。美国专利US2006/0047121A1中记载了“一种制备I型氯吡格雷硫酸氢盐的新方法”中涉及了将硫酸氢氯吡格雷盐溶于冰醋酸的溶液中然后加入反溶剂脂肪醚类溶剂混悬制得晶I型。这类方法避免了通过生成硫酸氢氯吡格游离雷碱的这一繁琐步骤。但同时也带来了制备方法的不稳定性。
在中国专利CN10050615C中记载了毛海舫等人发明的“一种制备I型氯吡格雷硫酸氢盐的方法”。其中,涉及了一种在硫酸氢氯吡格雷的游离碱中加入酮试剂,向酮溶液中滴加浓硫酸,其中在滴加浓硫酸过程中加入晶种,改善结晶效率,但难以实现对晶型质量的控制目的。
发明内容
本发明的目的是建立了两种直接使用氯吡格雷硫酸氢盐为原料通过制备过饱和溶液后采用沉淀或先种晶后沉淀方法制备获得高纯度晶I型,本发明使用的溶剂对环境友好,克服上述现有技术的多种不足。
为实现上述目的,本发明采用下述技术方案:
一种晶I型氯吡格雷硫酸盐的制备方法,包括以下步骤:
(1)在冰水冷却条件或室温条件下,将氯吡格雷硫酸氢盐溶解于正丙醇中,加热溶解并过滤得到澄清溶液;此处,冰水冷却温度可为5~10°C,室温条件可为15~30°C。
(2)往步骤(1)得到的澄清溶液中加入醚类沉淀溶剂,于20~37℃条件下搅拌6~9小时,得混悬溶液;
(3)将步骤(2)得到的混悬溶液过滤,得到固体样品,经真空干燥,得到晶I型氯吡格雷硫酸氢盐。
所述步骤(1)中氯吡格雷硫酸氢盐的质量与正丙醇的体积比为1:2~1:20(m/v,g/mL)。
所述步骤(2)中的醚类沉淀溶剂选自乙醚、叔丁基甲基醚或异丙醚。进一步优选,步骤(2)中的醚类沉淀溶剂为乙醚。
所述步骤(3)中的真空干燥温度为40~60℃,干燥时间为10~12小时;进一步优选为60℃干燥10个小时;
在同一发明构思下,向过饱和溶液中加入晶I型氯吡格雷硫酸氢盐晶种,可获得较宽的制备条件,其制备方法将如下所述。
另一种晶I型氯吡格雷硫酸盐的制备方法,包括以下步骤:
(1)在室温条件下,将氯吡格雷硫酸氢盐溶解于醇类有机溶剂中,加热溶解并过滤得到饱和澄清溶液;
(2)往步骤(1)得到的饱和澄清溶液中加入晶I型氯吡格雷硫酸氢盐晶种,通过搅拌或超声破碎晶种得到混悬溶液;
(3)往步骤(2)得到的混悬溶液中加入沉淀溶剂,经静置,过滤得到固体样品,在经40~60真空干燥,得到晶I型氯吡格雷硫酸氢盐。
所述步骤(1)中的醇类有机溶剂选自乙醇、正丙醇、乙腈、正丁醇或异丙醇。
所述步骤(1)中氯吡格雷硫酸氢盐的质量与醇类有机溶剂的体积比为1:2~1:20(m/v,g/mL)。
所述步骤(3)中使用的沉淀溶剂选自乙醚、石油醚、正己烷或环己烷。
本发明的切入点与上述文献和专利存在差异,表现为将氯吡格雷硫酸氢盐通过使用毒性较低的正丙醇有机溶剂制成饱和或过饱和溶液,加入沉淀溶剂方法迅速制备获得晶I型氯吡格雷硫酸氢盐,或通过使用多种有机溶剂将氯吡格雷硫酸氢盐完全溶解后,通过加入晶种后再使用多种有机沉淀溶剂制备获得晶I型氯吡格雷硫酸氢盐。
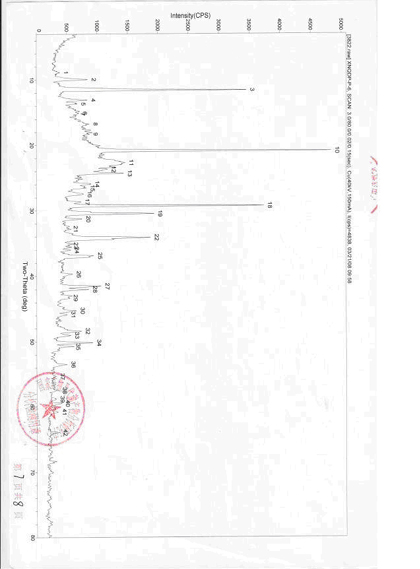
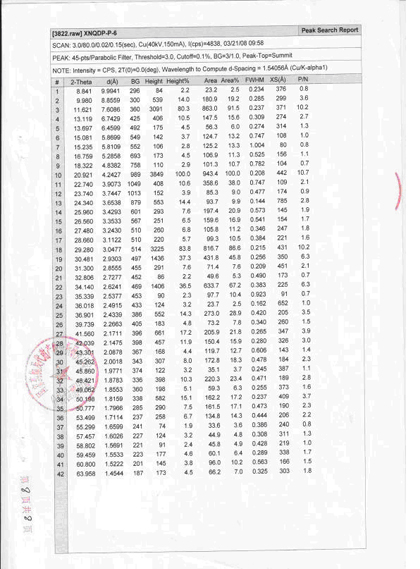
与现有技术相比,本发明的有益效果是:本制备方法工艺简单、操作容易,如在制备过程中加入晶种,可使工艺具备结晶快速、产品质量稳定(熔程短)、重现性高,通过对获得的晶I型氯吡格雷硫酸氢盐进行了粉末X射线衍射分析、DSC分析、红外光谱分析,结果表明本发明制备的晶I型属高纯度。氯吡格雷硫酸氢盐的晶I型与晶II型的粉末X射线衍射图谱在衍射峰数量、衍射峰位置、衍射峰强度等均存在明显差异(见图1、2);不同晶型的DSC图谱在吸热峰位置存在差异(见图3、4);红外光谱在3200~2000cm-1和1500~500cm-1区域的吸收峰存在较大差异(见图5、6)。
附图说明
图1a是晶I型氯吡格雷硫酸氢盐的粉末X射线衍射图谱。
图1b是晶I型氯吡格雷硫酸氢盐的粉末X射线衍射图谱数据。
图2a是晶II型氯吡格雷硫酸氢盐的粉末X射线衍射图谱。
图2b是晶II型氯吡格雷硫酸氢盐的粉末X射线衍射图谱数据。
图3是晶I型氯吡格雷硫酸氢盐的DSC图谱。
图4是晶II型氯吡格雷硫酸氢盐的DSC图谱。
图5是晶I型氯吡格雷硫酸氢盐的红外光谱图。
图6是晶II型氯吡格雷硫酸氢盐的红外光谱图。
具体实施方式
下面结合附图和实施例对本发明进一步说明。
实施例1
在20°C室温条件下,将1g氯吡格雷硫酸氢盐溶解于10mL正丙醇中,加热搅拌溶解并过滤得到澄清溶液;往溶液中加入250mL乙醚溶剂中速搅拌6小时后,真空抽滤得到白色固体,用50mL乙醚洗滤,得到固体样品,在60°C真空干燥12小时,得到0.65g晶I型氯吡格雷硫酸氢盐,熔点为183.5~184.5°C,收率65%。
实施例2
在10°C冰水冷却条件下,将1g氯吡格雷硫酸氢盐溶解于15mL正丙醇中,加热搅拌溶解并过滤得到澄清溶液;往溶液中加入370mL乙醚溶剂中速搅拌8小时后,真空抽滤得到白色固体,用50mL乙醚洗滤,得到固体样品,在60°C真空干燥12小时,得到0.7g晶I型氯吡格雷硫酸氢盐,熔点为183.5~184.5°C,收率70%。
实施例3
在20°C室温条件下,将0.8g氯吡格雷硫酸氢盐溶解于15mL正丙醇中,加热搅拌溶解并过滤得到澄清溶液;旋转蒸发抽干溶剂得到油状液体,往油状液体中加入150mL乙醚溶剂中速搅拌9小时,真空抽滤得到白色固体,用30mL乙醚洗滤,得到固体样品,在60°C真空干燥10小时,得到0.6g晶I型氯吡格雷硫酸氢盐,熔点为183.5~184.5°C,收率75%。
实施例4
在25°C室温条件下,将10g氯吡格雷硫酸氢盐溶解于100mL正丙醇中,加热搅拌回流并过滤得到澄清溶液;2~4滴/s的速度逐渐往溶液滴入500mL乙醚溶剂,中速搅拌9小时,真空抽滤得到白色固体,用100ml乙醚洗滤,得到固体样品,在60°C真空干燥12小时,得到8g晶I型氯吡格雷硫酸氢盐,熔点为183.5~184.5°C,收率80%。
实施例5
在20°C室温条件下,将0.5g氯吡格雷硫酸氢盐溶解于3mL正丙醇中,加热溶解并过滤得到饱和澄清溶液;往溶液中加入通过第一种方法制的I型氯吡格雷硫酸氢盐晶种约为10mg,通过搅拌或超声破碎晶种得到悬浮溶液;向得到的悬浮溶液中加入40mL石油醚溶剂,静置8小时,过滤得到固体样品,在60°C真空干燥,得到晶I型氯吡格雷硫酸氢盐,得到0.4g晶I型氯吡格雷硫酸氢盐熔点为183.5~184.5°C,收率80%。
实施例6
在25°C室温条件下,将1g氯吡格雷硫酸氢盐溶解于10mL正丙醇中,加热溶解并过滤得到饱和澄清溶液;往溶液中加入通过第一种方法制的I型氯吡格雷硫酸氢盐晶种约为20mg,通过搅拌或超声破碎晶种得到悬浮溶液;向得到的悬浮溶液中加入150mL石油醚溶剂,静置过夜,过滤得到固体样品,在60°C真空干燥,得到晶I型氯吡格雷硫酸氢盐,得到8.3g晶I型氯吡格雷硫酸氢盐熔点为183.5~184.5°C,收率83%。
说明书附图
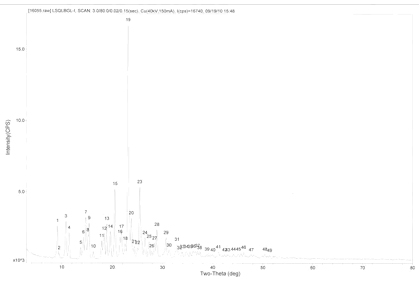
图1a
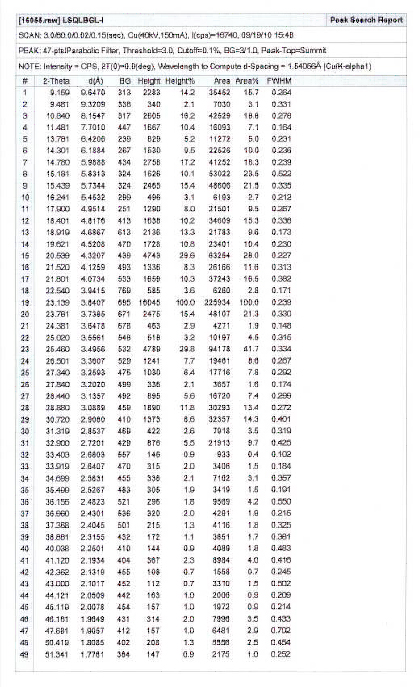
图1b
图2a
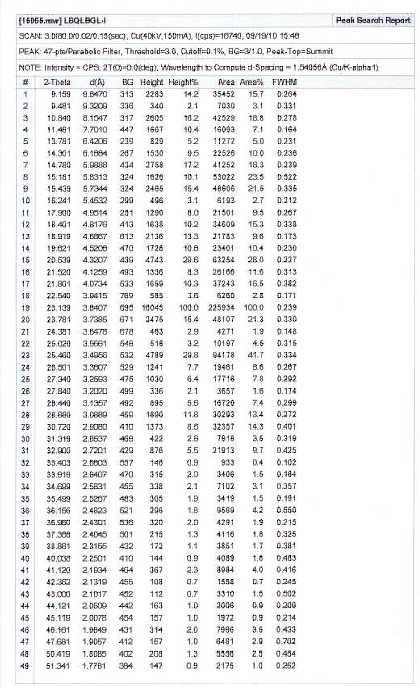
图2b
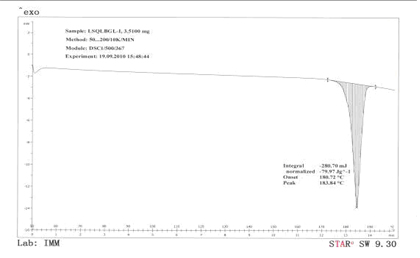
图3
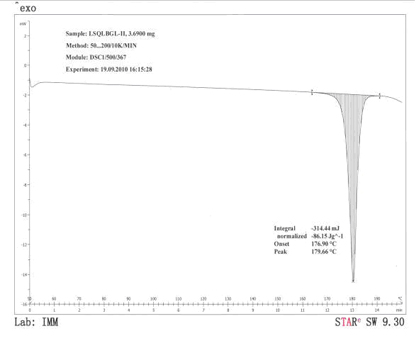
图4
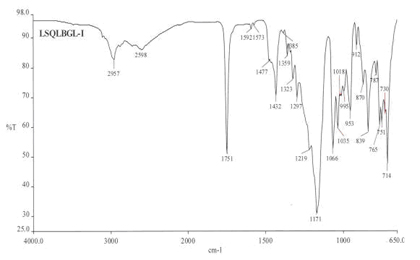
图5
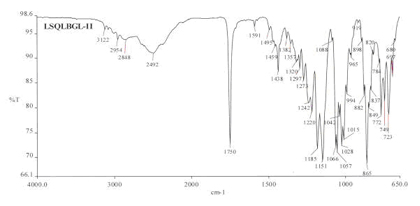
图6
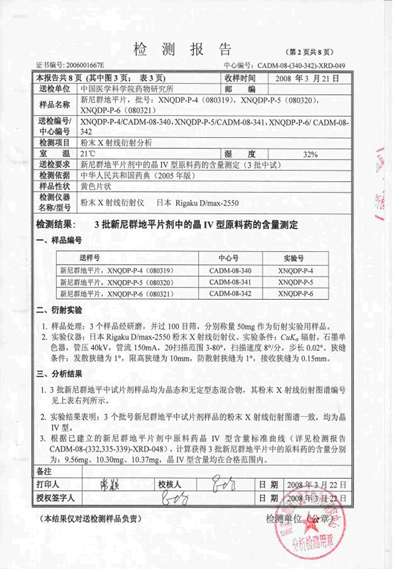
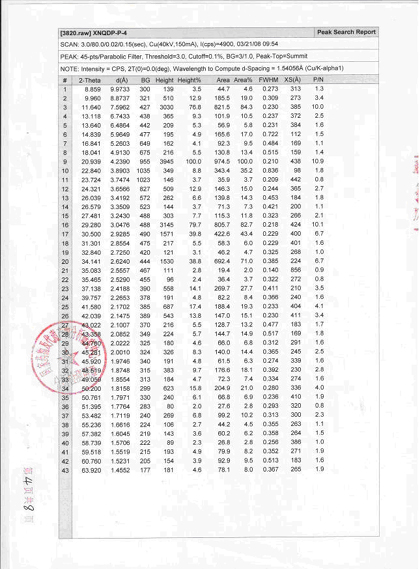
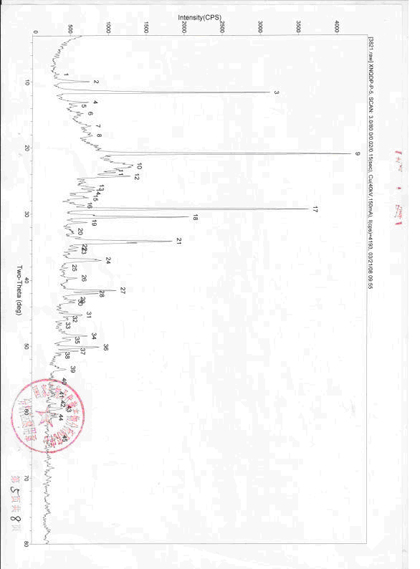
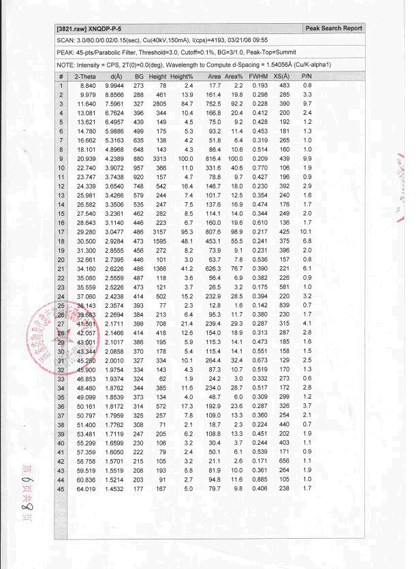
三、国家1.1类新药新尼群地平片中晶Ⅳ型检测报告